
National
Bureau
of
Standards
Library,
E-01
Admin.
Bldg.
FEB
3
1969
NBS
PUBLICATIONS
Allies
m
L.
014
z
T
NSRDS—NBS
19
°f%^
Ul02
'460H
Thermodynamic
Properties
Of
Ammonia as an
Ideal
Gas
NATIONAL BUREAU OF
STANDARDS

National Standard Reference Data
Series
National
Bureau of Standards
National
Standard
Reference
Data
System, Plan of
Operation,
NSRDS-NBS
1
—15 cents*
Thermal
Properties
of
Aqueous Uni-univalent
Electrolytes, NSRDS-NBS
2
—
45
cents*
Selected
Tables of Atomic Spectra, Atomic Energy
Levels and Multiplet Tables
—Si
H,
Si
hi.
Si iv, NSRDS-NBS
3,
Section
1
—35
cents*
Selected Tables of Atomic Spectra, Atomic Energy
Levels and Multiplet Tables—Si I,
NSRDS-NBS
3,
Section
2
—20
cents*
Atomic Transition Probabilities, Volume I, Hydrogen Through Neon, NSRDS-NBS
4
—
$2.50*
The Band
Spectrum of Carbon Monoxide, NSRDS-NBS
5
—70
cents*
Tables of Molecular Vibrational Frequencies, Part
1,
NSRDS-NBS
6
—
40
cents*
High Temperature Properties and Decomposition of
Inorganic Salts, Part
1,
Sulfates,
NSRDS-NBS
7—35
cents*
Thermal Conductivity of Selected Materials,
NSRDS-NBS
8
—
$1.00*
Tables of Biomolecular
Gas
Reactions, NSRDS-NBS
9
—
$2.00*
Selected Values of Electric Dipole Moments
for
Molecules
in the Gas
Phase, NSRDS-
NBS
10
—
40 cents*
Tables
of
Molecular
Vibrational
Frequencies,
Part
2,
NSRDS-NBS
11
—30
cents*
Tables
for
the Rigid Asymmetric Rotor:
Transformation
Coefficient for
Symmetric to
Asymmetric Bases and
Expectation Values
of
P'.
and
Pf
f
NSRDS-NBS
12
—60
cents*
Hydrogenation
of Ethylene
on Metallic
Catalysts,
NSRDS-NBS
13
—
$1.00*
X-Ray
Wavelengths and
X-Ray Atomic
Energy Levels,
NSRDS-NBS
14
—
40
cents*
Molten Salts: Electrical
Conductance,
Density
and Viscosity Data, NSRDS-NBS
15
—In
press
Thermal
Conductivity of
Selected Materials,
NSRDS-NBS
16
—
$2.00*
Tables
of Molecular
Vibrational
Frequencies, Part
3,
NSRDS-NBS
17
—30
cents*
Critical
Analysis
of the
Heat-Capacity of the
Literature and Evaluation of Thermodynamic
Properties
of Copper, Silver,
and Gold from
0 to 300
°K, NSRDS-NBS
18
—40
cents*
*
Send
orders with
remittance to:
Superintendent of Documents,
U.S.
Government Printing Office,
Washington,
D.C.
20402.
Remittances from
foreign countries
should include an additional one-fourth
of
the purchase
price
for
postage.

UNITED STATES
DEPARTMENT OF
COMMERCE
C.
R.
Smith,
Secretary
NATIONAL BUREAU OF
STANDARDS
•
A. V.
Astin, Director
Thermodynamic
Properties of
Ammonia
as
an Ideal Gas
Lester Haar
Institute for Basic Standards
National
Bureau
of Standards
Washington,
D.C.
20234
Reprinted from the
Journal of Research of the
National Bureau of Standards
A. Physics
and Chemistry
Vol. 72A, No.
2,
March-April
1968
NSRDS-NBS 19
National Standard
Reference Data Series-
National
Bureau
of Standards 19
Issued
August 1968
For sale by the Superintendent of
Documents,
U.S.
Government Printing
Office
Washington, D.C.,
20402
-
Price
20
cents


RATIONAL BUnSMJ
OF
STAH8A8C3
tiff
?
*
f
Foreword
The
National
Standard Reference
Data
System is a
Government-wide effort to provide for
the technical
community
of the United
States
effective access to
the quantitative data of physical
science,
critically
evaluated and
compiled for
convenience,
and readily accessible through a
variety of
distribution
channels. The System
was
established in
1963
by
action of the President’s
Office of
Science and
Technology and
the Federal
Council for Science
and Technology.
The responsibility to
administer the
System was
assigned to the
National Bureau of Standards
and an Office
of Standard
Reference Data was set up
at the
Bureau for this purpose.
Since
1963,
this Office
has developed
systematic
plans
for
meeting
high-priority needs for
reliable reference
data. It
has undertaken to
coordinate
and integrate existing
data evaluation
and compilation
activities
(primarily those under sponsorship
of Federal
agencies) into
a
comprehensive program,
supplementing
and expanding technical coverage
when
necessary, establishing and
maintaining
standards for the output
of the participating groups,
and providing mechanisms
for the dissemina-
tion of the output as
required.
The System now
comprises a complex of data
centers and other activities,
carried on in
Government
agencies, academic institutions, and
nongovernmental laboratories.
The independent
operational status of existing critical data projects is
maintained and encouraged.
Data centers
that are
components of the NSRDS produce compilations
of critically evaluated data,
critical
reviews
of the state of quantitative knowledge in specialized areas,
and computations of
useful
functions derived from
standard reference
data.
In addition, the centers and projects
establish
criteria for evaluation and
compilation
of data and make
recommendations on needed
modifications
or extensions of
experimental techniques.
Data publications of the
NSRDS
take a variety of physical
forms, including books,
pamphlets,
loose-leaf sheets and computer tapes. While most of the compilations have been issued by
the
Government
Printing
Office, several have appeared in scientific journals. Under
some circum-
stances,
private publishing houses are regarded
as appropriate
primary dissemination mechanisms.
The technical scope of
the NSRDS
is indicated
by
the principal categories of data
compilation
projects now active or being planned: nuclear
properties, atomic and
molecular properties, solid
state properties, thermodynamic and
transport properties, chemical kinetics,
colloid and surface
properties, and mechanical properties.
An important aspect of the
NSRDS is the advice and planning assistance which the National
Research Council
of
the National
Academy of Sciences-National
Academy of
Engineering provides.
These services
are
organized
under an overall
Review Committee which considers
the program
as
a whole and makes recommendations
on policy,
long-term
planning,
and international
collabora-
tion.
Advisory
Panels, each concerned
with
a
single technical
area, meet
regularly
to
examine
major portions
of
the
program, assign
relative priorities, and identify
specific
key problems in
need
of further attention.
For selected
specific
topics,
the Advisory Panels sponsor
subpanels
which make detailed studies of
users’ needs,
the present state of knowledge, and
existing data
resources as a basis for recommending
one or more
data
compilation
activities.
This assembly
of
advisory services
contributes greatly
to the guidance of NSRDS activities.
The NSRDS-NBS
series of publications
is intended primarily to include
evaluated reference
data and critical reviews
of
long-term
interest
to the scientific and
technical community.
A.
V. Astin,
Director.
Ill

Contents
PAGE
Foreword
III
1. Introduction 1
2. Partition function 2
3. Molecular
data
3
4. Thermodynamic
functions 5
5. Discussion 8
6. References
9
IV

Thermodynamic Properties of Ammonia
as
an
Ideal Gas*
Lester Haar
Institute for Basic Standards, National Bureau
of
Standards, Washington,
D.C. 20234
(December
15,
1967)
Thermodynamic functions for ammonia
as an
ideal
gas at one
atmosphere
pressure
have been
evaluated. The
contribution of the highly anharmonic out-of-plane vibrational mode, including its
large coupling
with rotation and its coupling with the other vibrational modes,
is
considered in detail.
Tables
of C°
P
IR,
(
H°
—
E°
0
)/RT
,
(
E°
0
—
G°)IRT
,
and S°/R have been calculated
at
closely
spaced inter-
vals from 50 to 5000 °K
within an overall uncertainty
of
less
than
0.1
percent
at
1000 °K.
Key Words: Ammonia, ideal
gas, thermodynamic functions.
1
.
Introduction
This
paper is part of a program originating with
the
Office of Standard Reference Data
of
the
National
Bureau
of Standards to obtain accurate thermody-
namic
properties for ammonia.
Subsequent papers of
this program
will include real-gas corrections
to the
ideal-gas properties
contained herein.
In this
paper the ideal-gas thermodynamic
properties
of ammonia in the
ground electronic state are calcu-
lated
at closely
spaced
temperature
intervals from
50 to 5000
°K. The calculations include
the contribu-
tions of
ordinary vibrational anharmonicity
and vi-
brational-rotational
coupling together with rotational
stretching
and rotational quantum
effects. Particular
attention
is given to the treatment
of
the
rotational
and
vibrational
anharmonic effects due to the
molecu-
lar inversion.
To this end a method
is developed for
calculating
this
contribution such that
an overall
accuracy
for the thermodynamic
properties
of am-
monia
approaching that of the
simplest
diatomic
molecules
is obtained.
In the
vibrational
ground state ammonia
is a pyram-
idal
molecule
with the
nitrogen atom
approximately
0.38 A
above the plane of the three
hydrogen
atoms.
The
distance between
the nitrogen atom
and each
of
the
hydrogen atoms is approximately 1.0
A.
Thus the
pyramid is relatively flat and the potential barrier to
vibration of the nitrogen
atom through
the
plane
of
the hydrogen
atoms
so
as
to
invert
the
pyramid
is
low
(approximately 2000 cm
-1
[l]).
1
Ammonia has
six vibrational
degrees
of freedom.
However, owing to symmetry
there are
only four
normal
vibrational
modes, two of which
have
double degen-
eracies. The
vibrational
structure may
thus
be char-
*This work was
supported in part by the Office of Standard
Reference Data, NBS.
1
Figures in brackets
indicate
the literature
references at the end of this paper.
acterized
by
the six quantum numbers
(viv
2
v£
3
v^),
where the
Vi
are the principal vibrational quantum
numbers associated with the vibrational modes
i
n
and
the /
characterize the small coupling of the de-
generate and V
4
asymmetric vibrational modes with
the angular momentum of the molecule. The V\ and
v
2
modes are symmetric, nondegenerate vibrations
corresponding to stretching the N-H bond and
spread-
ing the pyramid, respectively. (The v
2
mode is also
referred to as the out-of-plane vibration.)
The rotational
structure for the ground state is that of a symmetric
top, so
that the
rotational
energy levels are
charac-
terized
by
the
two
quantum
numbers
J
and K.
The v
2
mode
is the vibration
associated
with the
inversion of the
pyramid. Each
vibrational state
of this
mode is split into
two components,
corresponding
respectively
to symmetric
and antisymmetric
eigen-
functions.
(In the nomenclature
these
are identified
by
the
superscript
“
5
”
or “a” to the
quantum number
v
2 .)
The
spacing of these levels
is highly
anharmonic
except for
vibrational energies
sufficiently higher
or
lower
than the
potential barrier
to inversion. Thus
the
vibrational
levels for
the ground
state of the
v
2
mode
are only slightly
split,
of the order of a fraction
of a
wavenumber,
but the splitting
increases rapidly with
increasing
vibrational
quantum number. At
energy
levels
sufficiently elevated above
the barrier (greater
than
about 3000 cm
-1
)
the levels become uniformly
spaced with
a spacing of approximately half that
of
the v
2
fundamental. Similarly,
due to
inversion
the
rotational structure undergoes a transition from
that
of a pyramid
for the low values of
the
v
2
quantum
num-
ber to that of a planar molecule for the higher
values,
so that the rotational
motion is
also strongly
coupled
to the
v
2
mode. For a more detailed discussion of these
effects,
see
the review
by Herzberg
[2]
of the pyramidal
XY
2
molecule and
Nielsen
[3].
The energy structure
1

of the other vibrational modes is similar
to that of
typical, slightly anharmonic rotating vibrators.
In section
2 the equations for the
calculations are
developed. This is followed in section 3
by a discussion
of the molecular data, and in section 4
by a discussion
of the calculated properties and an evaluation
of their
accuracy. Finally, a discussion is given
in section 5
of some of the earlier work.
2. Partition Function
It is
convenient to represent the rotational and vi-
brational
structure by an expression for
which the
energy levels of the v
2
mode and its
coupling with
the other degrees
of freedom are written
explicitly.
Thus the
energy levels in
wavenumbers are written
T(
V,
y,
K) =G'{v) +F'(v,
J
,
K) +G
2
(v,
J,
K),
(1)
where the
primes denote that dependence on
v
2
is
omitted. The first
term in
eq
(1)
describes the
vibrational
energy
characterized by (viOvlfv!?)
and in
notation
similar to that
in Herzberg
[2]
is
approximated
by
G'(v)
=2
ViV
i
+
'*?
XijViVj
+
^'xu(vi—
l)vi
+
. . .
i i>j i
+
^34/3/4
+
^33(^3
—
^3)
+
g
44
(
l
\
—
V
4
)
+ . .
.
,
(2)
i,j^
2,
where the
Vi
are fundamentals and the x and
g
are
relatively small
anharmonicities. The second term in
eq
(1)
is
approximated
by
F'(vJ
,
K)
=
B'J{J
+
1) +
(C'
V
—B'
V
)K
2
(3)
-Dt'P(J
+ l)
2
-DrJU
+ I)K
2
-DK'K4
+
where
C
v
is
the rotational constant corresponding
to rotation about the symmetry axis
and
B
v
is the con-
stant for axes perpendicular
to
the symmetry axis.
The quantities D
J
V
,
D
J
V
K
,
and D%
are
small coefficients
associated
with centrifugal
distortion of the mole-
cule. The subscript
v to
each
of the coefficients of
eq
(3)
indicate dependence or coupling with the
vibrational motion.
The primes indicate that
de-
pendence on
V
2
is
omitted; that
is, the
rotational
constants of
eq
(3)
are evaluated
for
v
2
equal to zero.
The coupling of
the rotational
motion to
vibrations
other than
the
v
2
mode is
relatively
small and is
included
here only to first
order in the
fundamentals.
B'
V
=
B
0
—
^'
afvi
+
i
C'
v
=
Co-
a
c
t
Vi
+
,
i
#2
i
where B
0
and
Co
are
rotational
constants,
and
the a
are
small
corrections.
All
coordinates
involving
the
v
2
mode,
including
its
coupling
with the
other
degrees
of freedom
are included
in G
2
. Thus
G
2
includes
those
effects characterized
by
{v
x
v
2
v
l
Jv\
4
JK)
—
(vfivIfv^JK).
The partition
function
for the rotational
and
vi-
brational structure
may
be
written
Q=
2
exp{-T(v,J,
K)c
2
/T},
(4)
vJ,K
where T(v,
J,
K) is discussed in
eqs
(1),
(2),
and
(3)
.
C
2
is the second radiation
constant and
T
is the
absolute temperature. The sum
extends over all
rotational and vibrational
degrees of freedom in-
cluding
all
values of the
v,
J,
K
quantum numbers.
In the evaluation of
eq
(4)
the contributions
of the
vibrational anharmonicity and vibration-rotation
coupling effects contained in
eqs
(2)
and
(3)
are
all to be accounted for to first order;
that
is, only
terms linear in
x,
g
,
and a are retained. Though
G
2
is not conveniently
described
by
the usual
power
series in
the
vibrational
quantum numbers,
its con-
tribution, including coupling effects with the other
degrees of freedom, is also to be included to this order.
The contribution of the G
2
term is obtained
by
ex-
panding the partition function
about
the term-by-
term sum of the observed (Ot^OO) energy levels. This
summation incorporates the major effect of the highly
anharmonic
v
2
mode, which
is
the coupling
between
the symmetric
and
antisymmetric states, but it
does
not include the contribution of the v
2
coupling
with the
other
vibrational modes.
From
an examination of the
spectroscopic vibrational data (see Benedict et al.
[4]
),
it
can
be seen
that this
cross
coupling
produces a
shifting of energy levels of about the same magnitude
as
that of the other vibrational couplings (those repre-
sented by the x and
g
terms in
eq
(2)).
Thus the con-
tribution to
the
partition function of
this
cross coupling
should
be
of the same order as that from the other
anharmonicities, and a first order approximation for
this contribution should be adequate.
In the expansion that follows expressions
for the
cross coupling are developed and their contribution
for each
individual (Oi^OO)
state
obtained explicitly.
The cross coupling between a pair of vibrational states,
e.g.,
states corresponding to the particular
quantum
numbers
V\
and
v
2
,
is defined
as
G
Vl
v
2
— G(viVzOO)
—
G(yiOOO)
—
G(0
i>200)
,
(5)
where
G(v
iV
2
vfcv
l
f)
is the vibrational
energy. A useful
representation
for the coupling
involving the highly
anharmonic
v
2
mode
is the development of G
Vll>2
as a
power series
of products of the observed
energies,
that
is
(*viv
2
~
ki
2
G
(
1
^
1
000
)
•
G(0t>200)
F
....
(6)
The
constant
k\
2
is
readily determined in
terms of the
observed
coupling.
Thus for coupling
between the
fundamentals
the
observed coupling
xn
is given
by
2

*i2
=
Jti
2
C(1000)
•
G(0100)
+
. . .
=
k
i 2
uiv
2
+ ....
Note
that eq
(6)
reduces identically to the power series
of eq
(2)
when
the cross coupling
is between nearly
harmonic
modes.
Consider
the energy of the nearly harmonic
oscil-
lator
v
m
,
neglecting
all
anharmonicities
but
the cross
coupling
with v
2
. The energy levels
for such an oscil-
lator are
given
by
Gm
V
m
Vm
d
-
G
VmV
^.
Substituting
from
eq
(6)
and defining E
2
=
G(0z;200),
this
becomes
The prime indicates
that the
m
=
2 factor in the
product
corresponding to self
coupling of the
v>
mode is
omitted.
The coupling
of rotation
with
the v
2
mode is in-
cluded
by
evaluating
the classical
rotational partition
function
for each of
the
(CU2OO) energy
levels and em-
ploying these
as
separate
weighting factors to
each of
the terms
in
eq (12).
These
factors are the
usual ex-
pression for a classical
nonrigid rotator,
Qk{v
2)
=77
1/2
(cr!crc)~
1/2
(l
+
p
{1)
T
+p
(2)
r
2
).
(13)
The quantities c
r
B
and
<Jc
are given by.
Gm
—
VmVm
“1“
k
2
mVmVm
'
E
2
.
(7)
The
contribution of G
m
to
the partition function is the
factor
Qm
=
\
2
exp
—
[
(C
2
IT)
{VmVm +
klmE-zVmVm)]
(
8
)
where d
m
is the degeneracy of the
rath
mode. The
anharmonicity term in the exponential of eq
(8)
is now
expanded
and only the linear term retained to yield
Qm
=
C
2
VmVm
1
ij:
kzmVmVmEz
(
9
)
Using the identity
d_
da
2
ve~
the contribution
of
Q
m
becomes
<?m
=(l-e-r
(10)
where
=
cz
r
VC
J1
l?2
’
where B
V2
and
C
V2
are the observed rotational con-
stants for the particular (Ot^OO) energy level. The factor
1
+
p
(1)
T+
p
(2)
T
2
is the contribution of rotational
stretching (see, for example
(5
and
14)),
where the
p
are
calculated from the rotational
stretching
coeffi-
cients that
apply
to the particular
(0y
2
00)
level.
One further simplification is required in order to
evaluate
the out-of-plane contribution. This is to
obtain an approximation for the contribution
of the
energy
levels corresponding
to high values
of
the
v
2
quantum number. Note
that the v
2
mode is nearly
harmonic for energies
corresponding to
the fourth
quantum number and higher,
so
that the
contribution
of these levels may
be
replaced
by
the well-known
expression
for a slightly anharmonic rotating vibrator.
Combining
eqs
(12)
and
(13)
and including the closed
form approximation
for the contribution
of
the states
corresponding
to quantum numbers given
by v
> >
3
yields the expression
for
G>
(y,
K) used in this
calculation.
<?-={2|(§)
,
'V+P">r+^»r).
j
J
/2m=4yfe/2^/(e
Wra/T
-l).
(11)
exp
(
—
c->E
4
sIT
)
Bo/Co\
1/2
1
—
exp
(—
czv'zIT)
B
4
\Cy
The
factor
(1
—
f
2m
)
is
the
contribution
of
the inter-
action of
a
particular
(Ot^OO)
energy
state
with the
rath
vibrational mode,
and
it depends
explicitly
on
E
2
. It
is
clear then that
the
effect of
this
coupling is
to
modify each of
the
terms in
the
term-by-term
sum
over
the
((WX))
energy
levels
by
the
factor
(1
—
f
2m
)•
The
term-by-term
sum
including
cross
coupling with
the
other vibrational
modes
is
thus
given by
Q
V2
(vib)=
2) FI
0
~hm)
exp (—c
2
E
2
/T).
(12)
E
2
m
[]
(1
—
0
2
j)
(1
+
(f>
2
)
(l
+
p
{1)
T)
,
(14)
where E
=
E
2
and
the subscript to
E now refers to the
particular
v
2
quantum
number. The
quantity in the
braces
in
eq
(14)
includes
levels
only
up
to
t>2
=
3
a
.
Note
that
the rigid
rotational
contribution for the
ground
state has
been factored
from
eq (14).
In
the
0xp
(
—
c>E
JJ
1
)
second term the
factor
^ 7
4
,
,
^
is the har-
1
—
exp
(—
C2
v
2
l
1
)
monic oscillator
contribution
with
E
aS
as
the first level
289-015
0-68—7
3

3.
Molecular Data
and
v
2
the fundamental.
The factor 1
—
0
2
j
is the ordi-
nary
anharmonicity
coupling
with the yth vibrational
mode, where
Oij
p
Xjjdjdj
(exp (c-’Vi/T)
—
1)
(exp (c
2
VjlT)
—
1)'
Y
(xu
—
gal
3
)
di
(
di
+
1
)
(exp
(
czVi/T
)
—
l)
2
(15a)
The factor
1
+
f>i
is the well-known contribution of
rotation-vibration
coupling, where
4>i
=
di
(a?
1
«F\
\B
2
C)
(15b)
Details concerning the derivation of quantities in eqs
(15)
may
be
found in Friedman and Haar
[6],
Woolley
[7],
and Pennington and Kobe
[8],
for
example.
The contributions to
the partition function of the
two remaining terms G'
(
v
,
J
,
K
)
and F'
(v,
J
,
K) are
quite
straightforward
(see
[6, 7,
8]).
With
these
the
complete partition function is
finally obtained.
Mir
m
=
1
(i-«„)
[{'
(1
+
<M
isj
A:=l
'
Qtr
‘
Qqr
(16)
As noted earher the
primes
on the product summations
indicate that factors
containing the vibrational
coordi-
nate
1)2
are omitted.
The
quantity S
is the symmetry
number. For
the usual symmetric top molecule this has
the
value three. However, each of the components of
the
nondegenerate but split (Ot^OO) levels have been
counted
with
a
statistical weight
of unity, so
that the
factor
Q
V2
is larger
by a
factor
of two.
This
error is cor-
rected
by
setting
S
=
6.
(Incidentally
S
—
6
corre-
sponds to the symmetry for a planar configuration for
NH
3
,
and
eq
(16)
might
be
thought of
as
an expansion
about this
planar
configuration.) The product over m is
the harmonic
oscillator contribution for the modes
other than v
2
;
the products over
ij
and over k are the
corresponding anharmonicity
and rotation-vibration
coupling contributions, respectively. The
degeneracy
coefficients
d
are unity for m=
1,
2,
and two
for
ra
=
3,
4.
The
Oij
and
<\>k
are given
by eq
(15).
Qtr
is the usual
contribution of translation. Lastly,
the factor
Q
qr
is a
correction for rotational
quantum effects. It
is very
small
at temperatures for which
the vibrations are
appreciably
excited,
so
that it
is not coupled to the
v
2
mode. The contribution
Q
qr
is given
by
Stripp
and
Kirkwood
[9]
for an asymmetric rotator, which
for a
symmetric top rotator
reduces to
ob
Qqr
—
1
+
^
_
_
| |
B
4 cr
c)
15
I
-
4
o-c
32
The molecular data upon
which the
calculations are
based are
fisted in tables 1
and
2.
Table
1
fists the
data
sensitive
to
the inversion coordinates and utilized
in
the
term-by-term sum (the quantity in braces in
eq
(14)).
The vibrational energies corresponding to
i>
2
=
0,
3
are
from
[4].
The energies corresponding to
V
2
—
1,
2
are
from Garing et
al.
[10].
The values
fisted
for the
rota-
tional constants B
v
and
C
v
corresponding to
the
i>2
=
0 states are from
[4];
those
corresponding
to the
V
2
—
1,
2 states from
[10];
and to the
V
2
=
S states from
Benedict et al.
[11].
The rotational stretching constants
D{, D
J
V
K
,
D$ are the average of
the symmetric and
anti-
symmetric
values. The values for i>2
=
0
are from
[4];
for
V
2
—
1,
from
[10];
for
i>
2
=
3,
from
[11];
finally,
data
for
i)
2
=
2 were
taken equal to the
V
2
—I values.
Table 1. The
out-of-plane
molecular constants in cm~
1
,
\2
3,
(OV
2
OO)
State O'
0° 1* 1°
CVJ
CN
3*
3«
v
2
0.0 0.793 932.41 968.04 1597.42 1882.16 2383.46 2895.48
Br,
9.9443 9.939 10.07 9.89 10.26 9.73 9.50 9.20
Cv
2
6.196
6.198 6.05 6.13 5.90 6.13 6.16 6.26
&
c
2
8.1
x
10-'
8.9
x
10-*
8.9
x
10-'
-3.3x10-'
pjK
-
14.5
x
10-*
-
17.5
x
10-
4
—
17.5
x
10"*
15.0x10-*
D
K
7.5
x
10-"
10.5
X10-"
10.5
x
10-'
-11.3X
10-'
Table
2.
Vibrational, rotational and coupling constants in cm
-1
;
see eqs
(2),
(3)
t/,(l 0 0
0)
v'
t
=
Eia
—
Et,
i/
3
(0
0 1
0)
j/
4
(0
0 0
1)
3323.06
475 3443.381 1627.77
O
CO
1
II
H
*22
=
+
9
*33
=
—
18.5 *
44
=
8.8
*12
=
+ 20.6
*23
~
+
32.36
*34
=
—
17.3
£44
=+
2.65
*.
3
=-92
*24=-
10.7
*n
=
—
6.7
B,
=
9.35,
C
4
=
6.21
a?
=
0.135
of
=
+0.015
of
=
-F0.176
of
=-0.230
af=
.078 a£
=
+ 0.098 aj
=
—
0.009
af
=
+0.066
The remaining molecular data
are
given
in
table 2.
The vibrational fundamental
Vi
is
from Benedict
et al.
[12];
is from
[11];
V
4
is from
[10].
The constants
i^and
X
22
which correspond
to
the high
energy,
nearly-
harmonic
region of the V
2
mode are derived from the
reported
values for
the
(04*00) and
(05*00)
levels
at
3330 cm
-1
and 4315 cm
-1
given
by
Walsh and Warsop
[13]
and from theoretical considerations that at high
energies the spacing between the levels should become
nearly
equal to one-half the
(0100)
value. Also pertain-
ing
to this region are the rotational constants B
4
and C
4
which
are taken equal
to
the averages of the values
for
f2
=
3*
and 3
a
. The anharmonicity
coefficients X
12
,
X
13
,
X
14
,
X
23
,
X
24
,
X
33
,
and
X
34
are from
[4],
and
the coeffi-
cients
X
44
and
g
44
are from
[10].
The anharmonicity
coefficient
xn
is estimated from data
for similar
4
stretching
vibrations.
The coupling constants
af
are
from
[4].
The small coefficient
gw
is taken as zero.
The values used
for the atomic masses are
from
Cameron
and Wichers
[35J.
The values
used
for the
other
physical
constants
are given in
[36].
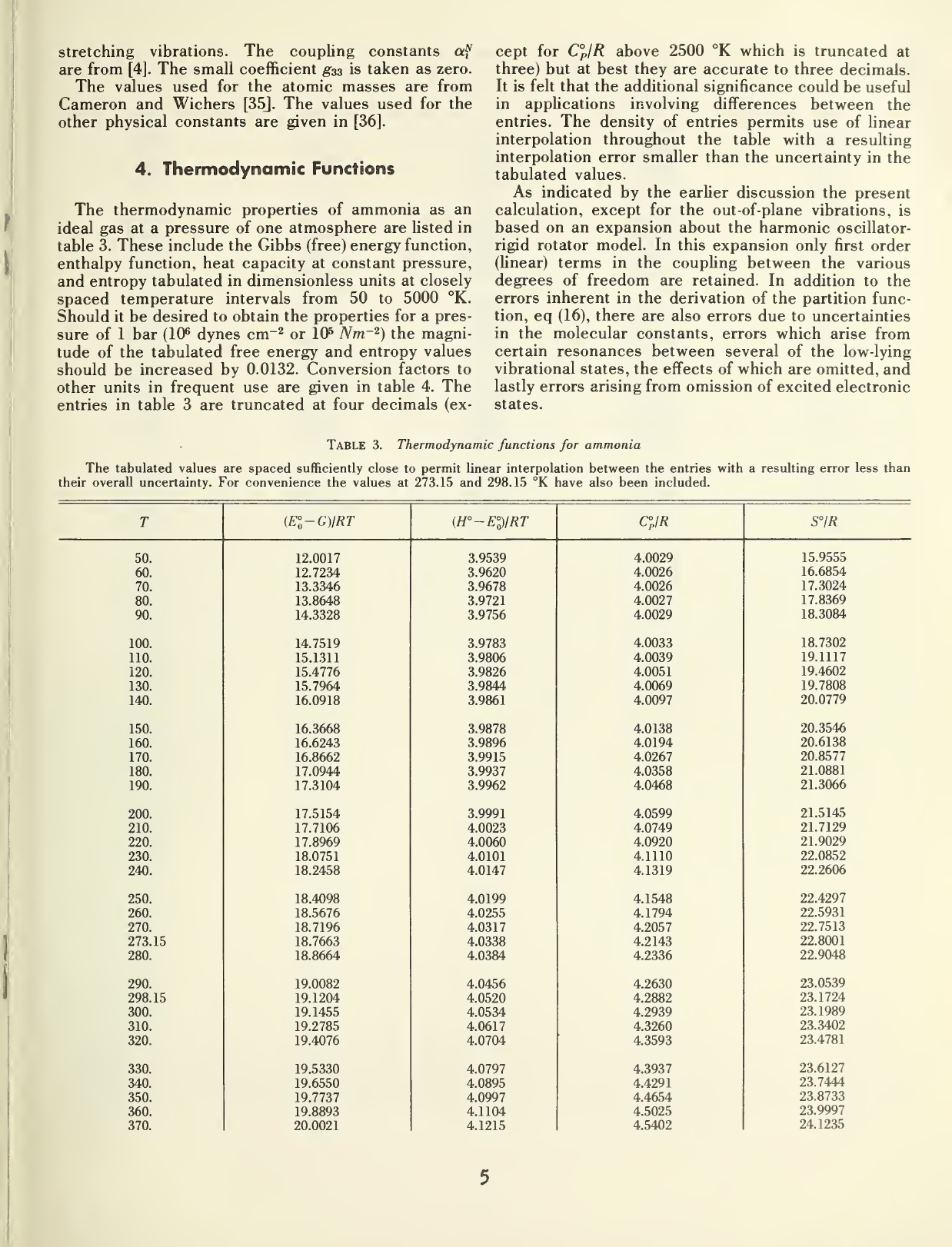
4.
Thermodynamic
Functions
The
thermodynamic
properties of
ammonia as an
ideal gas at a
pressure of one atmosphere are
listed in
table 3. These
include the
Gibbs (free) energy
function,
enthalpy
function, heat
capacity at constant
pressure,
and entropy
tabulated in
dimensionless units
at closely
spaced
temperature
intervals from 50 to
5000 °K.
Should it be
desired to
obtain the properties for a
pres-
sure of
1 bar
(10
6
dynes
cm
-2
or 10
5
Nm
~
2
)
the
magni-
tude
of
the tabulated free
energy and entropy
values
should be
increased by
0.0132. Conversion
factors
to
other
units in frequent use
are given in
table 4. The
entries in
table
3 are
truncated at
four decimals (ex-
cept for
C°p/R
above
2500 °K
which
is
truncated at
three) but at best they are
accurate to
three
decimals.
It
is
felt that the additional
significance
could
be
useful
in applications involving
differences
between
the
entries. The density of entries
permits
use of
linear
interpolation throughout the
table with
a resulting
interpolation error smaller than the
uncertainty
in
the
tabulated values.
As indicated
by
the
earlier
discussion the
present
calculation, except for the
out-of-plane
vibrations,
is
based on an expansion
about the
harmonic oscillator-
rigid rotator model. In this
expansion
only first order
(linear)
terms
in the coupling
between
the various
degrees of freedom
are retained. In
addition
to the
errors inherent
in
the
derivation of
the partition
func-
tion,
eq
(16),
there are also
errors
due to uncertainties
in the molecular
constants, errors
which arise from
certain resonances
between several
of the low-lying
vibrational states, the
effects of which are
omitted,
and
lastly errors arising from omission
of excited
electronic
states.
Table 3. Thermodynamic functions
for
ammonia
The tabulated values are spaced sufficiently close
to
permit linear interpolation between the entries with a
resulting error less than
their
overall uncertainty.
For convenience the
values
at 273.15 and
298.15
°K have also been included.
T
t*a
0
0
1
(H°-E°
0
)/RT Cp/R S°/R
50. 12.0017 3.9539
4.0029
15.9555
60.
12.7234 3.9620
4.0026
16.6854
70. 13.3346
3.9678
4.0026
17.3024
80. 13.8648
3.9721
4.0027
17.8369
90. 14.3328
3.9756
4.0029
18.3084
100. 14.7519
3.9783
4.0033
18.7302
110. 15.1311 3.9806
4.0039
19.1117
120. 15.4776 3.9826
4.0051
19.4602
130. 15.7964
3.9844
4.0069
19.7808
140. 16.0918
3.9861
4.0097
20.0779
150.
16.3668 3.9878 4.0138
20.3546
160. 16.6243 3.9896
4.0194
20.6138
170. 16.8662 3.9915
4.0267
20.8577
180.
17.0944 3.9937 4.0358
21.0881
190.
17.3104 3.9962
4.0468
21.3066
200. 17.5154
3.9991 4.0599
21.5145
210. 17.7106 4.0023
4.0749
21.7129
220.
17.8969 4.0060 4.0920
21.9029
230. 18.0751
4.0101 4.1110
22.0852
240. 18.2458 4.0147 4.1319
22.2606
250. 18.4098 4.0199 4.1548
22.4297
260. 18.5676 4.0255 4.1794
22.5931
270.
18.7196
4.0317
4.2057
22.7513
273.15 18.7663 4.0338 4.2143
22.8001
280. 18.8664 4.0384 4.2336
22.9048
290.
19.0082
4.0456 4.2630
23.0539
298.15 19.1204 4.0520 4.2882
23.1724
300.
19.1455 4.0534 4.2939
23.1989
310. 19.2785
4.0617
4.3260
23.3402
320.
19.4076 4.0704
4.3593
23.4781
330. 19.5330 4.0797
4.3937
23.6127
340.
19.6550 4.0895
4.4291
23.7444
350. 19.7737
4.0997
4.4654
23.8733
360. 19.8893 4.1104
4.5025
23.9997
370. 20.0021
4.1215
4.5402
24.1235
5

Table
3. Thermodynamic
functions
for
ammonia
—
Continued
T
(E°
0
-C)/RT
(H°-E°
t
)IRT C°JR
S°/R
380.
20.1121
4.1330
4.5785
24.2451
390.
20.2196
4.1449
4.6173
24.3646
400.
20.3247
4.1572
4.6565
24.4819
410.
20.4275
4.1699
4.6961
24.5974
420.
20.5282
4.1829
4.7359
24.7111
430.
20.6268
4.1962
4.7759
24.8230
440.
20.7234
4.2098
4.8161
24.9332
450.
20.8181
4.2237
4.8563
25.0419
460.
20.9111
4.2379
4.8966
25.1491
470.
21.0024
4.2524
4.9368
25.2548
480.
21.0921
4.2671
4.9771
25.3592
490.
21.1802
4.2820
5.0172
25.4622
500.
21.2669
4.2971
5.0572
25.5640
520.
21.4360
4.3278
5.1368
25.7639
540.
21.6000
4.3593
5.2158
25.9592
560.
21.7591
4.3912
5.2939
26.1503
580.
21.9137
4.4237
5.3712
26.3374
600.
22.0643
4.4566
5.4475
26.5208
620.
22.2109
4.4898
5.5230
26.7007
640.
22.3540
4.5232
5.5975
26.8772
660.
22.4937
4.5569
5.6711
27.0506
680.
22.6302
4.5907
5.7438 27.2210
700.
22.7638
4.6247
5.8157 27.3885
720.
22.8946
4.6588
5.8867
27.5533
740.
23.0227
4.6929
5.9568
27.7156
760.
23.1483
4.7271
6.0261 27.8754
780.
23.2715
4.7613
6.0946
28.0328
800.
23.3925
4.7954
6.1622
28.1879
820.
23.5113
4.8296
6.2291
28.3409
840.
23.6281
4.8637
6.2952 28.4918
860.
23.7430
4.8978
6.3604
28.6407
880.
23,8559
4.9317
6.4249 28.7877
900.
23.9672
4.9656
6.4885
28.9328
920.
24.0767
4.9994
6.5513 29.0761
940.
24.1845
5.0331
6.6133
29.2176
960.
24.2909
5.0667
6.6745
29.3575
980.
24.3957
5.1001
6.7348
29.4958
1000.
24.4990
5.1334
6.7943
29.6324
1020.
24.6010
5.1665
6.8530
29.7676
1040.
24.7017
5.1995
6.9108
29.9012
1060.
24.8010
5.2323
6.9678 30.0334
1080.
24.8991
5.2650
7.0239
30.1641
1100.
24.9960
5.2975 7.0792
30.2935
1120.
25.0918
5.3298
7.1335
30.4216
1140.
25.1864
5.3619 7.1870
30.5483
1160.
‘
25.2799
5.3938
7.2397
30.6738
1180.
25.3724
5.4256
7.2914 30.7980
1200.
25.4639
5.4571 7.3424
30.9209
1220.
25.5543
5.4884 7.3924
31.0427
1240.
25.6438
5.5195 7.4416
31.1633
1260.
25.7324
5.5504 7.4900 31.2828
1280.
25.8200
5.5811
7.5375
31.4011
1300.
25.9068
5.6115 7.5841 31.5183
1320.
25.9927
5.6418 7.6299 31.6345
1340.
26.0778
5.6718 7.6749 31.7495
1360.
26.1620
5.7016
7.7190 31.8636
1380.
26.2455
5.7311
7.7624
31.9766
1400.
26.3281
5.7604 7.8049 32.0886
1420.
26.4101
5.7895
7.8466
32.1996
1440.
26.4912
5.8184
7.8875
32.3096
6

Table
3.
Thermodynamic functions
for
ammonia
—
Continued
T
(E°
0
-C)IRT (.H°-E%)IRT
CUR
S°/R
1460.
26.5717
5.8470 7.9277 32.4187
1480.
26.6514
5.8754 7.9671
32.5268
1500. 26.7305 5.9035 8.0058
32.6340
1550.
26.9252
5.9729 8.0992
32.8981
1600. 27.1159 6.0407 8.1881
33.1566
1650.
27.3028 6.1071
8.2728 33.4099
1700.
27.4861 6.1720 8.3532
33.6581
1750.
27.6659 6.2354 8.4298 33.9013
1800.
27.8424 6.2974 8.5027
34.1398
1850.
28.0158 6.3579 8.5719 34.3737
1900.
28.1862 6.4171 8.6378 34.6032
1950.
28.3536 6.4748 8.7004
34.8284
2000.
28.5182 6.5312
8.7599
35.0494
2050. 28.6802
6.5863 8.8164 35.2664
2100.
28.8395
6.6400 8.8702 35.4795
2150.
28.9964 6.6925
8.9214
35.6889
2200.
29.1508 6.7437
8.9701
35.8945
2250.
29.3030
6.7937 9.0162 36.0966
2300.
29.4528
6.8425
9.0602
36.2953
2350.
29.6005
6.8901
9.1021
36.4906
2400.
29.7460
6.9366 9.1418 36.6826
2450. 29.8895
6.9820
9.1797 36.8715
2500.
30.0310
7.0263
9.216
37.0573
2550.
30.1706
7.0696 9.250 37.2402
2600.
30.3083
7.1118
9.282
37.4201
2650.
30.4441
7.1531
9.313
37.5972
2700.
30.5782
7.1933
9.342
37.7716
2750.
30.7106
7.2327
9.370
37.9432
2800.
30.8413
7.2711
9.397
38.1123
2850.
30.9703 7.3086
9.422
38.2789
2900. 31.0977
7.3452
9.445
38.4429
2950. 31.2236
7.3810
9.468
38.6046
3000.
31.3479
7.4160
9.490
38.7639
3100.
31.5922
7.4835
9.529
39.0757
3200.
31.8308
7.5480
9.564
39.3788
3300.
32.0640 7.6096 9.596
39.6736
3400. 32.2921 7.6684
9.623
39.9605
3500. 32.5152
7.7246 9.648
40.2398
3600. 32.7336 7.7783
9.670
40.5119
3700.
32.9474 7.8297 9.688
40.7771
3800.
33.1568 7.8788 9.704
41.0357
3900. 33.3621 7.9258
9.717
41.2879
4000.
33.5633 7.9707
9.728
41.5341
4100.
33.7607 8.0137 9.737
41.7744
4200.
33.9543
8.0548
9.743
42.0091
4300.
34.1443 8.0941 9.748
42.2384
4400.
34.3308 8.1317 9.750
42.4626
4500.
34.5140
8.1677 9.751
42.6817
4600.
34.6939 8.2021
9.749
42.8960
4700.
34.8706
8.2350 9.746
43.1056
4800.
35.0443 8.2664
9.741
43.3107
4900. 35.2151
8.2964 9.734
43.5115
5000.
35.3833
8.3253
9.725
43.7086
The
expansion about the harmonic oscillator limit
is
essentially
a low temperature expansion, and
the
errors
resulting
therefrom for
a
particular
vibrational
mode are small provided
j
is large, and are
of the
order of about the square of the linear term. The
7

Table 4
Conversion
factors
to
convert
tabulated value
to
quantity
having
dimensions
indicated
below
Multiply
by
Cal
mole
-1
°K
-1
1.98717
Cal
g
-1
°K
-1
0.116682
Joules
mole
-1
°K
-1
8.3143
Joules
g
-1
°K
-1
0.488197
largest
error of this type occurs for the v
4
mode. Thus
for the
most
sensitive function, the heat capacity, the
uncertainty at 2000 °K is less than 10
-2
dimensionless
units, but increases to the order of 0.1
at
5000 °K. The
higher
order anharmonicities
would
contribute an
un-
certainty
perhaps as large. Though the
anharmonicity
coefficients
Xn
and X13 have large uncertainties, the
errors introduced
by
these are
mitigated by
the
fact
that the fundamentals v\ and V3
are
large. A
50
percent
uncertainty in x
n
and
X13
results in
an
error of
about
0.05
dimensionless units in Cp/R at 5000
°K.
Setting
g33
equal
to zero introduces negligible error since this
coefficient
introduces a small
correction to an already
small
anharmonic contribution (owing to the large
value
of the V3 fundamental). The remaining anhar-
monicities (other than
the
*22
value in table
2)
are
probably
accurate to better than
several percent, and
the
uncertainties resulting
from these are compara-
tively
small. Uncertainties
arising from errors in the
rotational
fundamentals
are also comparatively small,
as are
uncertainties resulting from the
use of estimates
for some
of the rotational
stretching constants. The
uncertainties
that arise
from errors in the rotation-
vibration
coupling constants
are perhaps half as large
as those
arising from
errors in the anharmonicity
coefficients.
The
uncertainties
in the values
used for the funda-
mental
frequencies
are Jess than
several tenths of
a wavenumber,
and the uncertainty
from this source
is
negligible. Also
negligible are the uncertainties
due
to
errors
in the observed
overtones of the
r>
2
funda-
mental.
It
is somewhat
more difficult
to determine the un-
certainty
resulting
from
estimates
used
for the mo-
lecular
constants in the
second term in
eq
(14).
The
largest
uncertainty
here
arises from
possible errors in
v'
2
and E\? . An
uncertainty
of several
percent in these
results
in an
uncertainty
in Cp/R
of less than 0.002
dimensionless
units
at
1000
°K, which increases
to a
maximum
near
2000
°K of
less
than 0.01
dimensionless
units.
The
errors
from
estimates
of the
vibrational
coupling
constants
used in this
term
are
completely
negligible
below
2000
°K.
Even if a
100
percent uncer-
tainty
in their
contribution
is
assumed, the resulting
uncertainty
at
5000
°K
still
would
be less
than
0.1
dimensionless
units
in C
0
P
/R.
The
spectrum
of ammonia
is
further complicated
by
several
large
resonances
that
occur between some
of the
low
lying
vibrational
bands, such as the reso-
nance
between
the
combination
bands
(2^4
+
t^)
and
(vi
+
^3)
and between the
bands
2v
4
and v\
as discussed
in
[4].
Since the resonances tend to
displace the
re-
spective bands symmetrically
about their
unperturbed
values, it can be shown that the
errors are less
than
the neglected higher order anharmonicities.
A similar
type of
error occurs in the treatment
of
the
coupling
of the V2 mode with the other
modes. The coupling
con-
stants as reported are the average
of the symmetric
and
antisymmetric values. Thus the effect
of
this
approxi-
mation
is to introduce errors similar
to
those
arising
from neglect
of
the
resonances discussed above.
The
treatment
of
centrifugal
distortion, which includes
a
quadratic term in the
temperature, is consistent with
the other
approximations and the error from
this
source
at
5000
°K is probably less than 0.05 in Cp/R.
At the low temperature
end
there
is a small error
arising from the rotational quantum
approximation
()
qr
(eq
(17)).
At 100
°K this error is
less
than 10
-3
in Cp/R,
but
is
appreciably
larger
at 50 °K.
Finally, the U.V. absorption
spectrum indicates
bands extending
from
~
46,000
cm
-1
to
over
100,000
cm
-1
,
as
discussed,
for example,
in
[13].
The error
from omission
of these even
at the very
highest tem-
peratures
considered
here is
completely
negligible.
It is
felt, therefore, that
the uncertainty
in the tables
in the temperature
region
100
to
1000 °K is
probably
less
than 0.003
dimensionless units
in Cp/R.
This
uncertainty increases
to 0.02 at
2000 °K and
then in-
creases very
rapidly to
perhaps 0.3 at
5000 °K. A more
complete
list of the
uncertainty
estimates, including
those for the
enthalpy and
entropy, are
given in
table 5.
Table 5.
Estimates
of
uncertainty in dimensionless
units
T AC°
P
IR
A (H°-E°
0
)IRT
AS°/R
100
0.003
0.002
0.003
1000 .003
.002
.003
2000 .02
.01
.02
3000 .05
.02
.03
4000
.1
.05
.07
5000 .3
.1
.2
5.
Discussion
Ideal-gas calculations for
NH3 have been
reported
in the literature since the early
thirties, but
many of
these
calculations were
based
on relatively
incomplete
and inaccurate
molecular data and on
simplified
struc-
tural models. Also, for the most
part, the
temperature
ranges were
somewhat limited.
The references
[17-27]
are
representative of these
early efforts.
Some of the more recent
calculations
which have
been
widely
used
include those
of Harrison
and Kobe
[28]
who
calculated the heat
capacity, enthalpy,
and
entropy from
273.16
to
1500 °K.
They included
the
major effects associated
with the molecular
inversion
by summing the lower
states of the v
2
mode term
by
term, but omitted all vibration and
rotational
coupling
effects
other
than
rotational
stretching. The
review by
Davies reported in
the monograph
edited
by
Din
[29]
8

includes the specific heat,
entropy, and enthalpy from
200
to
1000
°K. The calculation follows
essentially the
procedure
in
[28],
except that
centrifugal stretching
is
omitted. Also it
appears that the contribution of the
states in the term-by-term
sum of the v
2
mode
was
omitted above the level
3
a
. The most
detailed of the
previous calculations is that
reported
by
Yungman
et
al.
[30],
(which also appears in
[16J),
in which the
free
energy, entropy,
and enthalpy are tabulated
at 100 °K
intervals from 298.15
to 6000
°K. In addition
to
the
term-by-term sum for the v
2
mode, this
work also in-
cludes several of the
first order
vibrational and rota-
tional coupling contributions.
It omits,
however, the
contributions
due
to coupling
of the v
2
mode
with the
.other vibrational modes,
and includes
a somewhat
simplified coupling of the v
2
mode with rotation.
These
calculations are based on
essentially the
same mo-
lecular
data as those reported
here,
except that the
values
used for the vibrational
coupling
constants
*i
3
and
*44
are
about twice as large.
One of the most
widely
used
set
of
tables
is that of JANAF
[31].
The reference
refers
to
the latest revision
for ammonia,
September
30,
1965. This
calculation follows the
procedures
of
[28]
and
[30],
though the
specific details are
not given in
the text. Finally, there
are several recent
calculations
based on somewhat simplified
molecular
models,
which
are included in references [32-34].
The figures 1 and 2 include
a comparison
of
some
of the previous calculations
with those made
here. The
ordinates are
the
deviations
from the present
calcula-
tions, that is the present calculation
minus the others.
Figure 1 is
a
plot of entropy
differences, and
figure 2
is a
plot
of heat capacity differences. It
is most inter-
esting
to
compare
the data of Yungman et al.
[30],
AS
R
0
I 2
3
4
5
TxlO
3
FIGURE
1.
Dimensionless entropy
differences.
Figure 2.
Dimensionless
heat capacity
differences.
curve
#5 in figure 1. At the low
temperatures the
agreement is quite
good, but as the
temperature
is
increased, a positive deviation
occurs until
about
3000 °K, when the deviation becomes
strongly
negative.
The
difference at the lower temperatures
is
probably
due to
their simplified
treatment of the coupling
of the
v
2
mode with rotation. At temperatures
above 1000
°K,
the dominant
contribution to the difference
is from
the
*24
term omitted in
[30]
which
yields
a positive
contribution. At
temperatures above 3000
°K,
the
dominant contribution is from
the omitted
X\
2
and
*23
terms
which yield large negative
contributions.
In
addition to these, the
high value they
used for
*
44
(larger by a factor of two) tends
to reduce the
differ-
ences at temperatures below 3000
°K, but
magnifies
them above. The same
general comments
apply
to the
JANAF
[31]
comparison as seen in curve
#1,
except
that the difference
goes negative at somewhat
higher
temperatures and the positive
deviations
are consider-
ably larger. Incidentally,
the JANAF and
Harrison
and
Kobe
[28
1
(curve
#4)
calculations
are
nearly identical
for temperatures
up to
1000
°K. Curve
#3 applies
to
a modified rigid
rotator harmonic
oscillator
calculation
as given in
an
older
(now superseded)
JANAF
table.
The
comparison for curve
#2 [29
1
illustrates
the sen-
sitivity
to
the
omission of the
hierarchy of
states
corresponding
to large
v
2
quantum
numbers
(
v
2
>
3).
The
differences plotted
in figure 2 exhibit
essentially
the same
behavior
as
those
in figure 1 except that
they
are
somewhat larger.
It
is a pleasure
to
thank
Joseph
Hilsenrath for many
helpful
technical discussions,
particularly relating
to the use of OMNITAB
to program
the calculation.
In this latter
connection, I also
wish to thank
Robert C.
Thompson.
6.
References
[1]
J.
D. Swalen
and
J.
A. Ibers,
J.
Chem. Phvs.
36,
1914
(1962).
[2]
G. Herzberg, Infrared
and Raman
Spectra of
Polyatomic
Mole-
cules, D. Van Nostrand
&
Co., New York
(1945).
[3]
H. H. Nielsen,
Revs. Modern Phys.
23
,
90
(1951).
[4]
W.
S.
Benedict
and E. K. Plyler,
Can.
J.
Phys.
35,
1235
(1957).
[5]
R.
S.
McDowell,
J.
Chem. Phys.
39,
526 (1963).
[6]
A.
S. Friedman and L. Haar,
J.
Chem. Phys.
22,
2051
(1954).
[7]
H. W. Woolley,
J.
Res. NBS
56,
105 (1956).
[8]
R. E. Pennington
and
K. A.
Kobe,
J.
Chem. Phys.
22,
1442
(1954).
[9]
K. F. Stripp and
J.
G. Kirkwood,
J.
Chem. Phys.
19,
1131
(1951).
[10] J.
S. Garing, H. H. Nielsen, and
K. N. Rao,
J.
Mol.
Sped.
3,
496
(1959).
[11]
W.
S. Benedict, E. K. Plyler,
and E.
D. Tidwell,
J.
Chem. Phys.
29,
829
(1958).
[12]
W.
S.
Benedict,
E. K. Plyler, and E. D. Tidwell,
J.
Chem. Phys.
32,
32 (1960).
[13]
A. D. Walsh
and
P.
A. Warsop, Trans.
Faraday
Soc.
57,
345
(1961).
[14]
Y.
S. Yungman, Zhur. Fiz. Khim.
35,
319 (1961).
[
15
j
I. N. Godnev, Zhur. Fiz. Khim.
27,
1702 (1953).
[16]
V. P.
Glushko, Ed., Termodinamicheskie
Svoistva
Individ-
ual’nykh Vestchestv,
II, Acad.
Sci., USSR, Inst, of Applied
Chemistry,
Moscow
(1962).
[17]
D.
S.
Villars, Phys. Rev.
38,
1552
(1931);
J.
Am.
Chem.
Soc.
53,
2006 (1931).
[18]
M. Trautz,
Ann.
Physik
9
,
465 (1931).
9

[19]
F. A. F.
Schmidt, Forsch.
Arb.
Ing. Wes.
8,
91
(1937).
[20]
R. Overstreet
and
W.
F. Giauque,
J.
Am.
Chem. Soc.
59
,
254
(1937).
[21]
K. K. Kelley,
U.S.
Bur. Mines, Bull.
407 (1937).
[22]
E.
Justi,
Spez-Warme, Enthalpie
und
Dissociation Technischer
Gase,
J.
Springer, Berlin
(1938).
[23]
C. C. Stephenson and H. O. McMahan,
J.
Am. Chem.
Soc.
61,
437 (1939).
[24]
K.
S.
Pitzer,
J.
Chem.
Phys.
7,
251 (1939).
[25]
R. F. Haupt and E. Teller,
J.
Chem. Phys.
7,
925 (1939).
[26]
H. Zeise, Z.
Elektrochem.
47,
380
(1941).
[27]
H. W. Thomson, Trans. Faraday
Soc.
37,
344 (1941).
[28]
R. H. Harrison and K. A. Kobe, Chem. Eng. Progress
49,
351
(1953).
[29]
F. Din, Thermodynamic Functions of Gases
1
(Butterworths,
London,
1956).
[30]
Y.
S.
Yungman,
L. V. Gurvich, and N. P. Rtischeva, Tr. Gos.
Inst.
Prikl. Khim.
49
,
26
(1962).
[31]
JANAF
Thermochemical Tables, Distrib.
by
Clearinghouse
for
Federal
Scientific
Information,
PB168370-1,
Sept.
30,
1965.
[32]
C.
L. Mader,
Ideal
Gas
Thermodynamics of Detonation Prod-
ucts, U.S.
A.E.C., AECU-4508
(1959).
[33]
V. N.
Khlebnikova and
V. P. Morozov, Ukrain. Khim. Zhur.
24, 3
(1958).
[34]
S.
Sundaram, F. Suszek,
and F. F. Cleveland,
J.
Chem. Phys.
32
,
251
(1960).
[35]
A.
E. Cameron and E.
Wichers,
J.
Am. Chem. Soc.
84,
4192
(1962).
[36]
NBS Tech.
News Bull.
47,
No.
10
(Oct. 1963).
(Paper 72A2-495).
10

Announcement of
New
Publications in
National Standard Reference Data
Series
Superintendent
of Documents,
Government Printing
Office,
Washington,
D.C.
20402
Dear
Sir:
Please
add my name to the announcement list of new publications to be issued
in the
series: National Standard Reference
Data
Series
—
National Bureau of
Standards.
Name
Company..
Address....
City
(Notification key
N-337)
State
Zip
Code


NATIONAL
BUREAU OF
STANDARDS
The National
Bureau of
Standards
1
was
established by an act
of Congress
March
3,
1901. Today, in addition to
serving as
the
Nation’s central
measurement
laboratory,
the Bureau is a principal
focal point in
the
Federal Government for
assuring
maxi-
mum application of the
physical and
engineering
sciences
to
the
advancement of tech-
nology in industry
and commerce. To
this end
the Bureau conducts
research and
provides central national services
in three
broad program areas and provides
cen-
tral national services in a fourth.
These are:
(1)
basic
measurements and standards,
(2)
materials measurements
and standards,
(3)
technological
measurements and
standards, and
(4)
transfer of technology.
The Bureau
comprises the Institute
for Basic
Standards, the
Institute for
Materials
Research, the Institute
for
Applied Technology, and
the Center for
Radiation Research.
THE INSTITUTE FOR BASIC STANDARDS provides the
central basis
within
the
United
States
of
a
complete
and consistent
system of physical measurement, coor-
dinates that
system with the measurement
systems of other nations, and furnishes
essential
services leading to accurate and
uniform physical measurements
throughout
the Nation’s scientific community, industry, and
commerce.
The Institute consists
of an Office of Standard Reference Data and a group of divisions organized
by
the
following
areas of science and engineering:
Applied
Mathematics
—Electricity
—
Metrology—
Mechanics
—
Heat
—Atomic
Phys-
ics
—
Cryogenics
2
—
Radio
Physics
2
—
Radio
Engineering
2
—
Astrophysics
2
—Time
and
Frequency.
2
THE INSTITUTE FOR MATERIALS
RESEARCH
conducts materials
research lead-
ing to methods, standards
of measurement, and data needed
by industry, commerce,
educational institutions,
and government. The Institute
also provides advisory
and
research services
to other government agencies. The
Institute consists
of an Office of
Standard
Reference
Materials
and a group of divisions organized
by the following
areas of materials
research:
Analytical Chemistry—
Polymers—
Metallurgy
—
Inorganic Materials
—
Physical
Chemistry.
THE INSTITUTE
FOR
APPLIED TECHNOLOGY
provides
for
the creation of appro-
priate
opportunities for
the use and
application of technology within the Federal Gov-
ernment
and
within the civilian sector of
American
industry. The primary
functions
of the Institute
may be broadly classified as
programs
relating to technological
meas-
urements and
standards and techniques for the transfer of technology. The
Institute
consists of
a Clearinghouse for Scientific and Technical Information,
3
a
Center for
Computer
Sciences and Technology, and a group of technical divisions and offices
organized
by the following fields of technology:
Building Research—Electronic Instrumentation
—
Technical
Analysis
—
Product
Evaluation
—Invention and Innovation
—
Weights and
Measures
—
Engineering
Standards
—
Vehicle
Systems
Research.
THE
CENTER
FOR
RADIATION RESEARCH
engages in research,
measurement,
and
application
of radiation
to the solution of Bureau mission problems and the
problems
of
other agencies
and institutions. The Center for Radiation Research con-
sists of
the following
divisions:
Reactor
Radiation
—Linac Radiation
—
Applied Radiation
—
Nuclear
Radiation.
1
Headquarters
and Laboratories
at Gaithersburg, Maryland, unless
otherwise noted; mailing
address Washington,
D. C. 20234.
2
Located
at
Boulder,
Colorado
80302.
3
Located
at 5285
Port
Royal
Road,
Springfield,
Virginia 22151.

U.S.
DEPARTMENT
OF COMMERCE
WASHINGTON, D.C. 20230
POSTAGE AND
FEES PAID
U.S.
DEPARTMENT OF
COMMERCE
OFFICIAL
BUSINESS
